Produkter uten medisinsk formål
Publisert:
|
Oppdatert:
Endringer
På denne siden finner du informasjon om hva produkter uten medisinsk formål er og hvilke krav som gjelder for disse produktene.
Innhold på siden
Regelverket for medisinsk utstyr gjelder også for lignende produkter som ikke har et medisinsk formål, for eksempel kontaktlinser uten styrke og injeksjonsprodukter. At disse produktene skal oppfylle en rekke krav til sikkerhet og ytelse gjør dem tryggere å bruke.
Hva er produkter uten medisinsk formål?
Det finnes en rekke produkter som ligner medisinsk utstyr i funksjon og risikoprofil, men som ifølge produsenten bare har et estetisk eller et annet ikke-medisinsk formål, såkalte vedlegg XVI produkter. Eksempel på slike produkter er fargede kontaktlinser uten styrke, injeksjonsprodukter, kosmetiske implantater og laser til fjerning av tatoveringer.
Produktene er listet opp i forordning (EU) 2017/745 (MDR) vedlegg XVI og omfatter følgende produktgrupper:
-
Kontaktlinser uten styrke, for eksempel fargede kontaktlinser.
-
Produkter beregnet på å bli helt eller delvis innført i menneskekroppen ved hjelp av en kirurgisk invasiv metode med henblikk på å endre anatomien eller fiksere kroppsdeler, bortsett fra tatoveringsprodukter og piercinger. Dette er for eksempel implantater med kosmetisk formål.
-
Stoffer, kombinasjon av stoffer eller produkter beregnet på utfylling av ansiktet, huden eller slimhinner ved hjelp av subkutan, submukøs eller intradermal injeksjon eller annen innføring, bortsett fra dem beregnet på tatovering. Dette er for eksempel fillere til ansiktet.
-
Utstyr beregnet på å redusere, fjerne eller ødelegge fettvev, for eksempel utstyr til fettsuging, lipolyse eller lipoplastikk.
-
Utstyr som avgir elektromagnetisk stråling med høy intensitet (f.eks. infrarødt, synlig lys og ultrafiolett lys) beregnet på bruk på menneskekroppen, herunder koherente og ikke-koherente kilder, monokromatiske og bredspektrede kilder, for eksempel lasere og utstyr med intenst pulserende lys, til hudforbedring, tatovering eller hårfjerning eller annen hudbehandling.
-
Utstyr beregnet på stimulering av hjernen ved bruk av elektrisk strøm eller magnetiske eller elektromagnetiske felt som trenger inn i kraniet, for å endre nevronal aktivitet i hjernen. Dette kan for eksempel være produkter til hjernestimulering med strøm eller elektromagnetiske felter, f.eks. til å stimulere hukommelse eller konsentrasjon.
Hvilke krav gjelder for produkter uten medisinsk formål?
I henhold til MDR artikkel 1 nummer 2 vil kravene i MDR også gjelde for produktene uten medisinsk formål oppført på listen i vedlegg XVI. I forordningen omtales begge produktgruppene som «utstyr». Dette vil si alle krav i forordningen som stilles til «utstyr» gjelder for både medisinsk utstyr og produktene uten medisinsk formål.
Les mer om krav til utstyr her.
I tillegg til kravene som følger av MDR skal produktene oppfylle kravene i forordning (EU) 2022/2346, som gir felles spesifikasjoner for produkter uten medisinsk formål.
Produktene skal klassifiseres etter klassifiseringsreglene i MDR, men det gjelder særlige klassifiseringsregler for de produktene uten medisinsk formål som er aktive. Disse reglene finnes i forordning (EU) 2022/2347.
Særlige krav til merking
I de felles spesifikasjonene for produkter uten medisinsk formål stilles det særlige krav til sikkerhetsinformasjon som skal følge med produktet. Blant annet stilles det krav til at noen av produktene kun skal håndteres av helsepersonell eller lege, og at de ikke skal brukes på personer under 18 år. Informasjon om aldersgrensen skal tydelig markeres på produktet og gjelder blant annet for implantater, injeksjonsprodukter og utstyr beregnet på å redusere, fjerne eller ødelegge fettvev.
I likhet med medisinsk utstyr skal produkter uten medisinsk formål CE-merkes.
Aktørroller og deres forpliktelser
Kravene og forpliktelsene til de ulike markedsdeltakerne er de samme for produkter uten medisinsk formål som for medisinsk utstyr. Det er viktig å avgjøre hvilken rolle man har for å finne hvilke krav man må oppfylle. Les om krav og forpliktelser for de ulike markedsaktørene.
Oversikt over de ulike rollene for markedsdeltakere
| Markedsdeltaker | Definisjon i MDR artikkel 2 og IVDR artikkel 2 |
|---|---|
| Produsent | en fysisk eller juridisk person som framstiller eller helrenoverer utstyr, eller som får utstyr designet, framstilt eller helrenovert, og som markedsfører nevnte utstyr i eget navn eller under eget varemerke. |
| Autorisert representant | enhver fysisk eller juridisk person etablert i Unionen* som har fått og har akseptert en skriftlig fullmakt fra en produsent plassert utenfor Unionen, til å utføre bestemte oppgaver på dennes vegne med hensyn til dennes forpliktelser i henhold til denne forordning. |
| Importør | enhver fysisk eller juridisk person etablert i Unionen* som bringer utstyr fra en tredjestat i omsetning i Unionen*. |
| Distributør | enhver fysisk eller juridisk person i omsetningskjeden, utenom produsenten eller importøren, som gjør utstyr tilgjengelig på markedet fram til ibruktaking. |
| Helseinstitusjon | en organisasjon som har som hovedformål å pleie eller behandle pasienter eller fremme folkehelsen. |
* Med «Unionen» menes her EU/EØS-området og Tyrkia.
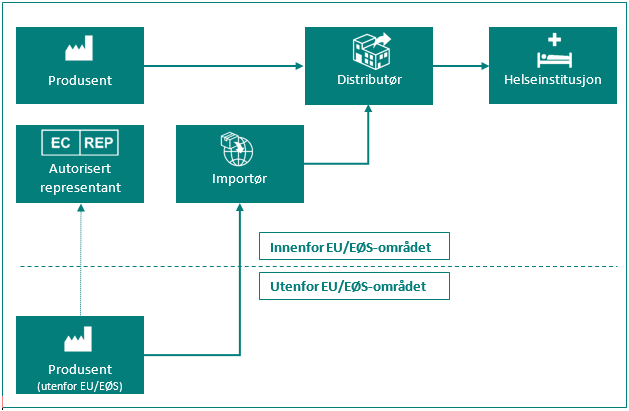
Figur: Markedsdeltaker i omsetningskjeden.
Registering
Eudamed er den europeiske databasen for medisinsk utstyr, denne er ennå ikke obligatorisk å bruke. Frem til Eudamed er obligatorisk å bruke vil en del av kravene i MDR bortfalle, blant annet kravet til registering i Eudamed.
DMP oppfordrer norske produsenter, autoriserte representanter, importører og sammensettere av systemer og prosedyresett til å registrere seg også før registreringsplikten blir obligatorisk.
Følgende aktører kan registrere seg i Eudamed:
-
Produsenter
-
Autoriserte representanter
-
Importører
-
Sammensettere av systemer og prosedyresett
Hvis man vil registrere seg i Eudamed, gjør man dette på EU-kommisjonens hjemmeside.
Det er foreløpig ingen nasjonale krav til registering av produkter uten medisinsk formål i Norge.
Markedsovervåking og meldeplikt for hendelser
Produsenter av produkter uten medisinsk formål skal ha et system for overvåking etter at utstyret er plassert på markedet. Systemet skal være en del av kvalitetssystemet. Les mer om overvåking etter at utstyr er plassert på markedet her.
Overgangsordninger for produkter uten medisinsk formål
I tillegg til de overgangsreglene som følger av MDR finnes det egne overgangsregler for produkter uten medinsk formål. Disse overgangsreglene fremgår av forordning (EU) 2022/2346 og gjelder produkter
-
hvor det må gjennomføres klinisk utprøving,
-
hvor fremgangsmåten for samsvarsvurdering krever medvirkning fra et meldt organ,
-
som er omfattet av et sertifikat i samsvar med direktiv 93/42/EØF.
Som følge av endringer i overgangreglene i MDR er overgangsreglene i forordning (EU) 2022/2346 endret ved forordning (EU) 2023/1194.
EU-kommisjonen har utarbeidet et Q&A-dokument om overgangsordningene for produkter uten medisinsk formål. Klikk her for å komme til dokumentet.
Under finner du ufyllende informasjon om hver enkelt overgangsordning.
Overgangsordning for produkter hvor det må gjennomføres klinisk utprøving
Et produkt hvor produsenten har til hensikt å utføre, eller er i ferd med å utføre, en klinisk utprøving for å bekrefte samsvar med de generelle kravene til sikkerhet og ytelse, og samsvarsvurderingen krever involvering av et meldt organ, kan bringes i omsetning eller tas i bruk frem til 31. desember 2029, forutsatt at følgende vilkår er oppfylt:
-
Produktet er lovlig omsatt i EU/EØS før 22. juni 2023 og fortsetter å oppfylle kravene som gjaldt for produktet før 22. juni 2023, og
-
produktets design og tiltenkte formål må ikke være vesentlig endret
Formålet med overgangsregelen er å gi produsentene tilstrekkelig tid til å utføre de nødvendige kliniske utprøvingene og framgangsmåten for samsvarsvurdering. Av denne grunn vil overgangsregelen opphøre dersom produsentene ikke innen nærmere angitte frister har innledet de kliniske utprøvingene eller framgangsmåten for samsvarsvurdering:
-
Fra 22. juni 2024 til 22. desember 2024 kan produktet bare bringes i omsetning eller tas i bruk dersom sponsoren har mottatt en underretning fra den berørte medlemsstaten i samsvar med MDR artikkel 70 nr. 1 eller 3.
-
Fra 23. desember 2024 til 31. desember 2027 kan produktet bare bringes i omsetning eller tas i bruk dersom sponsoren har startet den kliniske utprøvingen.
-
Fra 1. januar 2028 til 31. desember 2029 kan produktet bare bringes i omsetning eller tas i bruk dersom det meldte organet og produsenten har underskrevet en skriftlig avtale om utføring av samsvarsvurderingen i henhold til MDR vedlegg VII nr. 4.3 andre avsnitt.
Overgangsordning for produkter hvor fremgangsmåten for samsvarsvurdering krever medvirkning fra et meldt organ
Et produkt hvor prosenten produsenten ikke har til hensikt å utføre en klinisk utprøving, men der et meldt organ skal medvirke i samsvarsvurderingen, kan bringes i omsetning eller tas i bruk frem til 31. desember 2028, forutsatt at følgende vilkår er oppfylt:
-
Produktet er lovlig omsatt i EU/EØS før 22. juni 2023 og fortsetter å oppfylle kravene som gjaldt for produktet før 22. juni 2023, og
-
Produktets design og tiltenkte formål må ikke være vesentlig endret
Formålet med overgangsregelen er å gi produsentene tilstrekkelig tid til å utføre samsvarsvurderingen. Av denne grunn vil overgangsregelen opphøre dersom produsenten ikke innen nærmere angitt frist har innledet framgangsmåten for samsvarsvurdering:
-
Fra 1. januar 2027 til 31. desember 2028 kan produktet bare bringes i omsetning eller tas i bruk dersom det meldte organet og produsenten har underskrevet en skriftlig avtale om utføring av samsvarsvurderingen i henhold til MDR vedlegg VII nr. 4.3 andre avsnitt.
Overgangsordning for produkter som er omfattet av et sertifikat i samsvar med direktiv 93/42/EØF
Forordning (EU) 2017/745 (MDR) fastsetter overgangsregler for produkter med sertifikat utstedt i samsvar med MDD:
Sertifikater utstedt fra 25. mai 2017 som fortsatt var gyldige 26. mai 2021 og som har utløpt før 20. mars 2023 skal anses som gyldig i henhold til datoene nevnt i artikkel 120 nr. 3a, forutsatt at én av følgende krav er oppfylt:
- Produsent og meldt organ har signert en skriftlig avtale i samsvar med MDR vedlegg VII nr. 4.3 andre avsnitt før sertifikatets utløpsdato. Dette gjelder for produktet omfattet av det utgåtte sertifikatet eller et produkt som er ment å erstatte dette produktet.
- Kompetent myndighet i EU/EØS har innvilget et unntak fra samsvarsvurderingsprosedyren i henhold til MDR artikkel 59 nr. 1, eller har påkrevd produsenten å utføre gjeldende samsvarsvurderingsprosedyre i henhold til artikkel 97 nr. 1.
For produkter hvor disse vilkårene ikke er oppfylt introduserer forordning (EU) 2022/2346 egne overgangsregler for produkter uten tiltenkt medisinsk formål:
Et produkt som omfattes av MDD hvor sertifikatet har utløpt etter 26. mai 2021 og før 20. mars 2023, kan bringes i omsetning eller tas i bruk frem til
- 31. desember 2027 for produkter i risikoklasse III og for implanterbare produkter i risikoklasse IIb, med unntak av suturer, agraffer, tannfyllingsmidler, tannbøyler, tannkroner, skruer, kiler, plater, tråder, stifter, klemmer og forbindelsesledd, og
- 31. desember 2028 for produkter i risikoklasse IIb som ikke er nevnt ovenfor, produkter i risikoklasse IIa, samt produkter i risikoklasse I som er brakt i omsetning i steril tilstand eller har en målefunksjon
Overgangsregelen forutsetter at
- produktet fortsetter å oppfylle kravene i MDD,
- produktets design og tiltenkte formål ikke er vesentlig endret,
- produktet ikke utgjør en uakseptabel helse- og sikkerhetsrisiko for brukere eller andre aspekter av vernet av folkehelsen,
- produsenten innen 24. mai 2024 har et kvalitetssystem i samsvar med MDR artikkel 10 nr. 9,
- produsenten eller den autoriserte representanten innen 26. mai 2024 har sendt en formell søknad til et meldt organ i samsvar med MDR vedlegg VII nr. 4.3 første avsnitt, og
- produsenten og det meldte organet innen 26. september 2024 har signert en skriftlig avtale i samsvar med MDR vedlegg II, nr. 4.3 andre avsnitt.
Kravene i MDR til overvåking etter at utstyret er brakt i omsetning, markedstilsyn, sikkerhetsovervåking, registrering av markedsdeltakere og utstyr får vil få anvendelse i stedet for de tilsvarende kravene i MDD. Det meldte organet som har utstedt sertifikatet skal fortsette å være ansvarlig for overvåking etter at utstyret er brakt i omsetning, med mindre produsenten har inngått avtale om slik overvåking av utstyret med et meldt organ utpekt etter MDR.
Senest 26. september 2024 skal det meldte organet produsenten har signert en skriftlig avtale med være ansvarlig for overvåking av utstyret omfattet av denne avtalen.
Ordningene for overføring av overvåking fra det meldte organet som utstedte sertifikatet til det meldte organet utpekt i samsvar med artikkel 42, skal være klart definert i en avtale mellom produsenten og det meldte organet utpekt i samsvar med artikkel 42, og der det er praktisk mulig, det meldte organet som utstedte sertifikatet. Det meldte organet utpekt i samsvar med artikkel 42 skal ikke være ansvarlig for samsvarsvurderingsaktiviteter utført av det meldte organet som utstedte sertifikatet.
Mer informasjon
DMP gjennomførte høring av det nye regelverket i mars 2023.
Den norske oversettelsen av forordning (EU) 2022/2346 (PDF) om felles spesifikasjoner for produkter uten medisinsk formål er tilgjengelig på lovdata. Her finner du også norsk oversettelse av forordning (EU) 2022/2347 (PDF) om omklassifisering av grupper av aktivt utstyr uten et tiltenkt medisinsk formål.
På Kommisjonens nettsider finnes det flere veiledninger om produkter uten medisinsk formål:
- MDCG 2023-6 om demontrasjon av ekvivalens
- MDCG 2023-5 om kvalifisering og klassifisering
- Q&A om overgangsordningene for produkter uten medisinsk formål