Field Safety Corrective Action
Published:
Changes
Manufacturers, authorized representatives, importers, and distributors have a legal obligation to report or communicate field safety corrective actions
Page contents
What is a Field Safety Corrective Action?
A field safety corrective action is an action that manufacturers can implement based on technical or medical reasons to prevent or reduce the risk of serious incidents when using a medical device that has been placed on the market.
When systematic errors or deficiencies are found, or other considerations affecting the safety of the medical device in use, the manufacturer shall implement safety corrective actions(Field Safety Corrective Action – FSCA). The purpose may be to rectify errors, inform users about proper and safe use, and minimize the risk of serious incidents when using the device. The safety corrective safety action is communicated to users through a safety notice (Field safety notice – FSN).
Examples of Field Safety Corrective Actions
- Withdrawal of a medical device from the market
- Modifications to a device, temporary or permanent, such as component replacement or changes to user manuals, software updates
- Information about changes in maintenance
- Recommendations regarding device usage, calibration needs, or based on the analysis of patient results
- Changes in sample or reagent storage
Responsibilities of Manufacturers and Authorized Representatives (AR)
The manufacturer should assess the need for field safety corrective actions based on its risk management system. Both the manufacturer and AR are obliged to report field safety corrective actions to the responsible authority in the country where the manufacturer or AR has its business address. Norwegian manufacturers should report their FSCAs to the Norwegian Medical Products Agency.
Reports should also be sent to the authorities in all affected countries in the EU/EEA where the relevant medical device is on the market. If the device is certified by a notified body, the notified body should also be informed.
Using a safety notice, the manufacturer must ensure that users of the relevant medical device are informed about the field safety corrective action implemented.
How to Report a Corrective Safety Measure
Responsibilities of Importers and Distributors
Importers and distributors must collaborate with the manufacturer to ensure that necessary field safety corrective actions are taken for a mredical device they have imported/distributed. If relevant, this collaboration can also include AR and responsible authorities.
Importers and distributors must:
- Ensure that the field safety corrective action is implemented according to the manufacturer's plan
- Distribute the manufacturer's safety notices to their users or the next level in the supply chain
- Keep track of the implementation of the measure in their market
- Maintain a register of equipment covered by safety notices
- Notify the manufacturer (and AR if applicable) if they assess or have reason to believe that equipment they have circulated has faults or deficiencies affecting the equipment's safety
- Notify the manufacturer if they receive complaints or reports from healthcare professionals
Responsibilities of Healthcare Facilities (Users and Owners)
The safety notice describes how users and owners should handle the safety corrective action and how the information should be disseminated.
Find more information on handling safety notices as a healthcare facility.
Responsibilities of Private Individuals
If you, as a private individual, become aware of a field safety corrective action related to a medical device you use, follow the instructions provided, or alternatively, contact the entity that supplied the medical device.
Role of the Norwegian Medical Products Agency with Field Safety Corrective Actions
It is the manufacturer's responsibility to ensure that the field safety corrective action is implemented as planned. The Norwegian Medical Products Agency's task is to assess the received report on the safety corrective action and monitor its implementation. The Norwegian Medical Products Agency evaluates whether the manufacturer's corrective safety measure is sufficient and if there is a need for changes or additional corrective measures.
The Norwegian Medical Products Agency collaborates with other authorities in the EU to monitor safety corrective actions. When a Norwegian manufacturer reports a safety corrective action to the Norwegian Medical Products Agency, this information is shared with the European network so that other authorities in the countries where the medical device is on the market are informed. This is in addition to the manufacturer's own notification to the relevant authorities.
EU regulations on medical devices
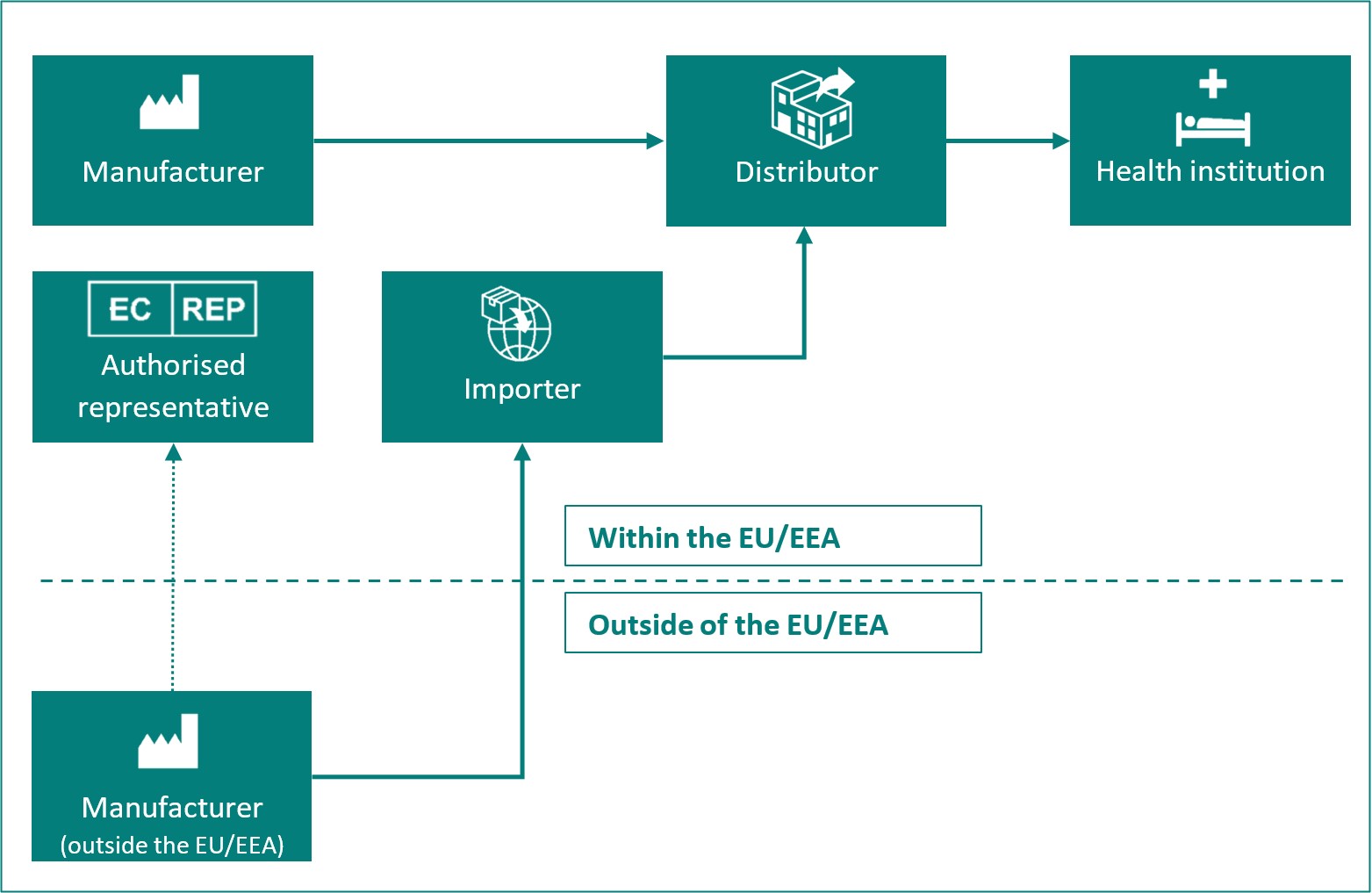
Overview of the different roles of economic operators:
| Economic operator |
Definition in MDR Article 2 and IVDR Article 2 |
| Manufacturer | A natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trademark. |
| Authorised representative | Any natural or legal person established within the Union* who has received and accepted a written mandate from a manufacturer, located outside the Union*, to act on the manufacturer's behalf in relation to specified tasks with regard to the latter's obligations under this Regulation. |
| Importer | Any natural or legal person established within the Union* that places a device from a third country on the Union* market. |
| Distributor | Any natural or legal person in the supply chain, other than the manufacturer or the importer, that makes a device available on the market, up until the point of putting into service. |
| Health institution | An organisation the primary purpose of which is the care or treatment of patients or the promotion of public health. |